
The server flowchart
The server combines softwares and scripts developed by the MIG team with well established bioinformatic tools.
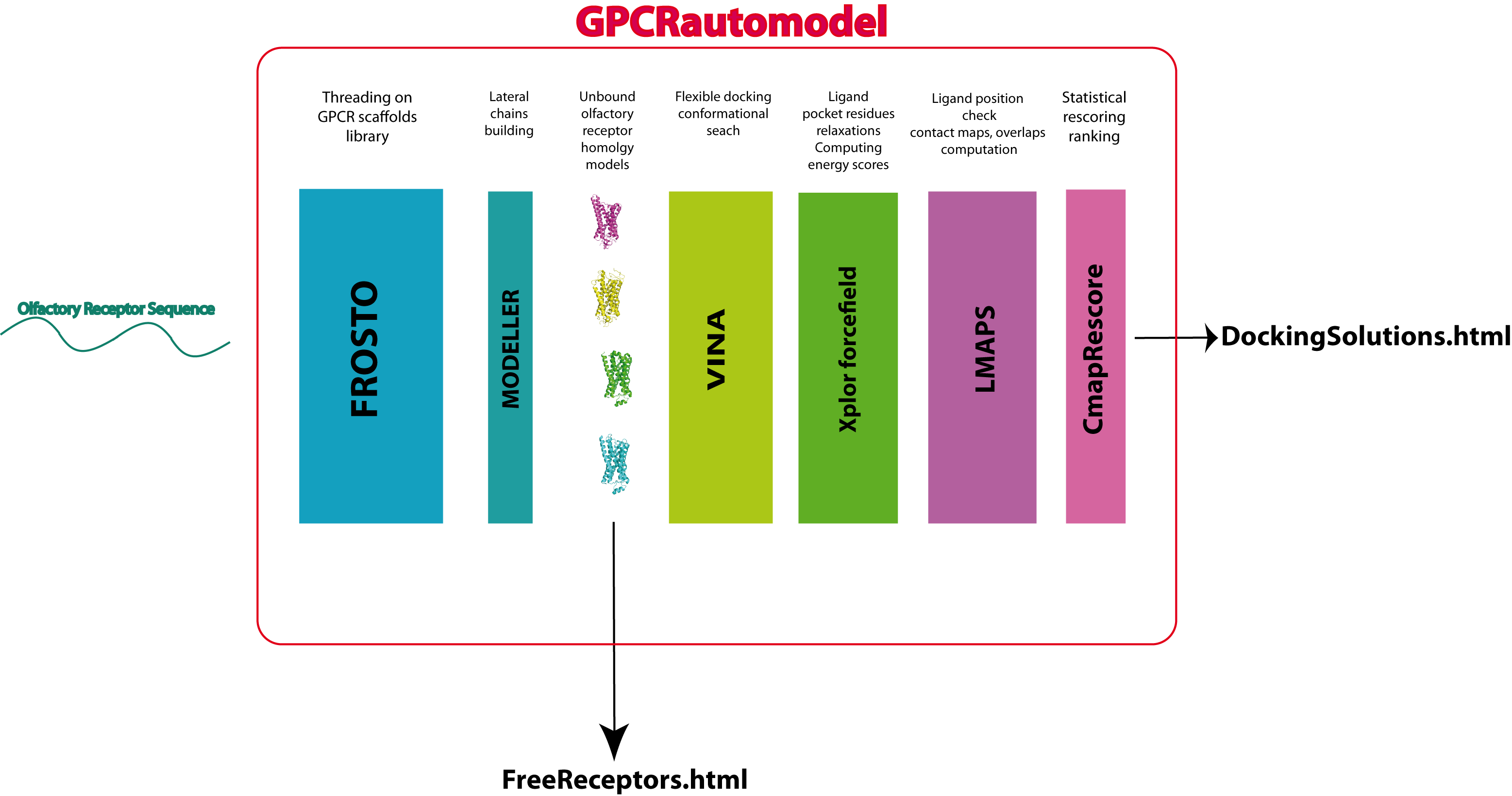
Modeling the structure of a query protein
The method used to predict the fold of any submitted query sequence is a homology-based approach. A multiple alignment of the query is constructed using BLAST. Because the homology relationship can be weak among GPCR proteins, convergence is rarely reached. It is therefore necessary to carefully parse the blast output in order to retrieve fragments of sequences with too many gaps. The submitted query fasta is also treated by PSIPRED. The sequence is checked for the existence of at least 7 helices, so be carefull GPCRautomodel could potentially deny you from modeling your sequence at this stage!
The query sequence is then aligned onto the selected template structure(s) using the threading software FROSTO. Threading methods are intended to detect and align remote homologs, i.e., homologs for which the sequences have undergone multiple substitutions, insertions and deletions. Such sequences often have less than 20% sequence identity and are difficult to align using ordinary sequence comparison methods. To overcome this problem, threading methods make use of the 3D-structure of the template proteins. The knowledge of the 3D-structure allows the algorithm to take into account residues that are in close contact in the structure although they may be far in the sequence. This additional piece of information is often critical to obtain accurate alignment, especially when the sequence identity between the protein sequences is low.
The current GPCRautomodel database of structures incorporates six distinct class-R GPCR.
| β2adrenergic | bovine rhodopsin | β1adrenergic | human A2A-adenosine | Chemokine | D3 Dopaminergic | |
| β2adrenergic | 2.04 | 0.79 | 2.12 | 2.24 | 1.35 | |
| bovine rhodopsin | 22.4 | 2.07 | 2.49 | 2.19 | 1.74 | |
| β1adrenergic | 71.0 | 22.2 | 1.96 | 2.44 | 1.68 | |
| human A2A-adenosine | 33.3 | 22.6 | 37.2 | 2.66 | 2.27 | |
| Chemokine | 25.4 | 24.0 | 28.3 | 22.5 | 1.80 | |
| D3 Dopaminergic | 37.6 | 27.9 | 39.4 | 29.9 | 20.5 |
The upper triangular part of this table displays the root-mean-square deviations (rmsd) of the Cαs after optimal superimposition of the seven transmembrane helices (TMHs) using VAST. The lower triangular part displays the sequence identity in the seven TMHs after the structural alignment.
The backbone coordinates of the model will be tranfsered from the template structure using the alignment provided by FROSTO. The lateral chains of the model will be constructed using MODELLER.
A confirmation email is sent to the user at job completion. It provides an html link to a results page where all models can be interactively manipulated in 3D using the JMOL applet.
Predicting the binding mode of a ligand molecule
The binding prediction process can be schematically divided into three separate stages:
- The VINA software is used to dock the ligand molecule on each model independantly.
- The XPLOR protocole is used to minimize the energy of the ligand-receptor docking complexes
- The LMAPS module is used to compute the ligand-receptor fingerprint in the minimized energy conformation.
The statistical scoring scheme implemented in LMAPS accounts for the stability of the conformational space vicinity elements in order to ponderate the energy of each conformation. A confirmation email is sent to the user upon docking and scoring completion. The predicted binding modes can be interactively vizualized in a fashion similar to the free models.
Illustration of the binding mode predictions
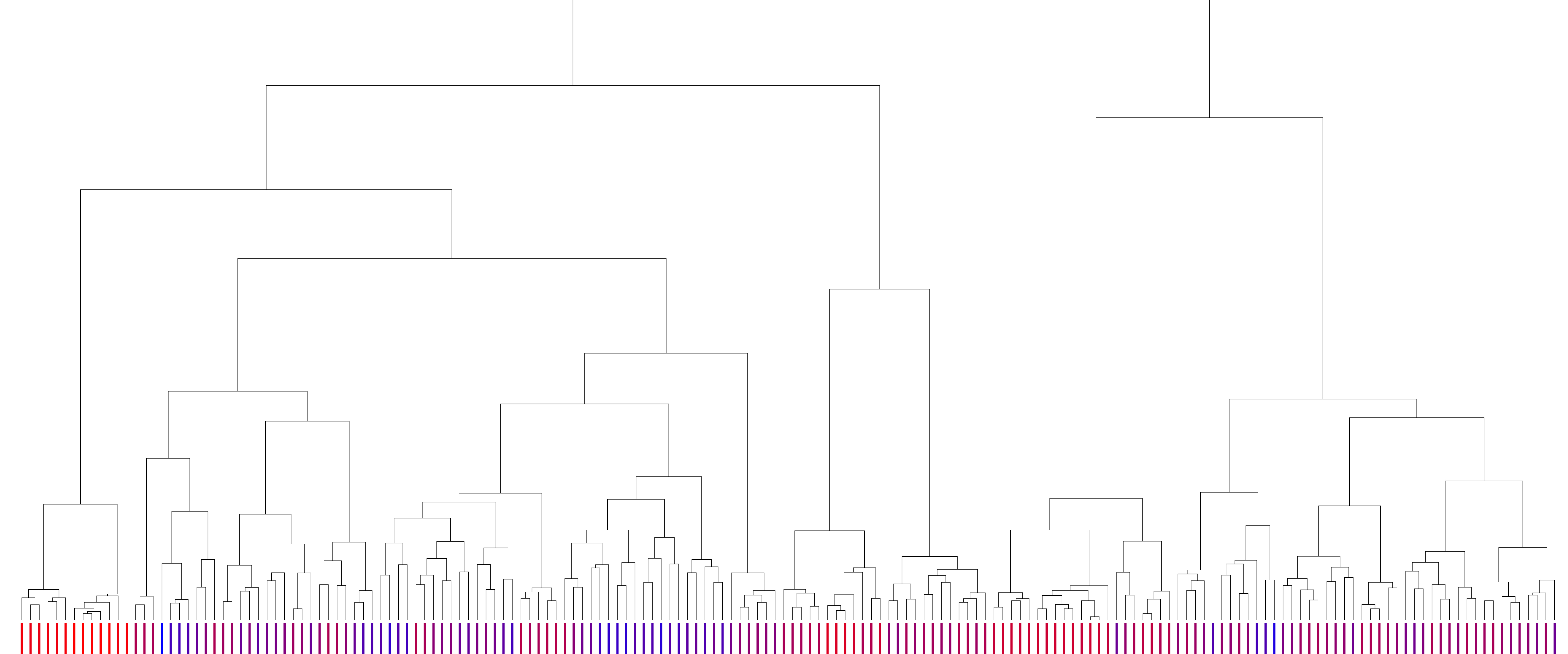
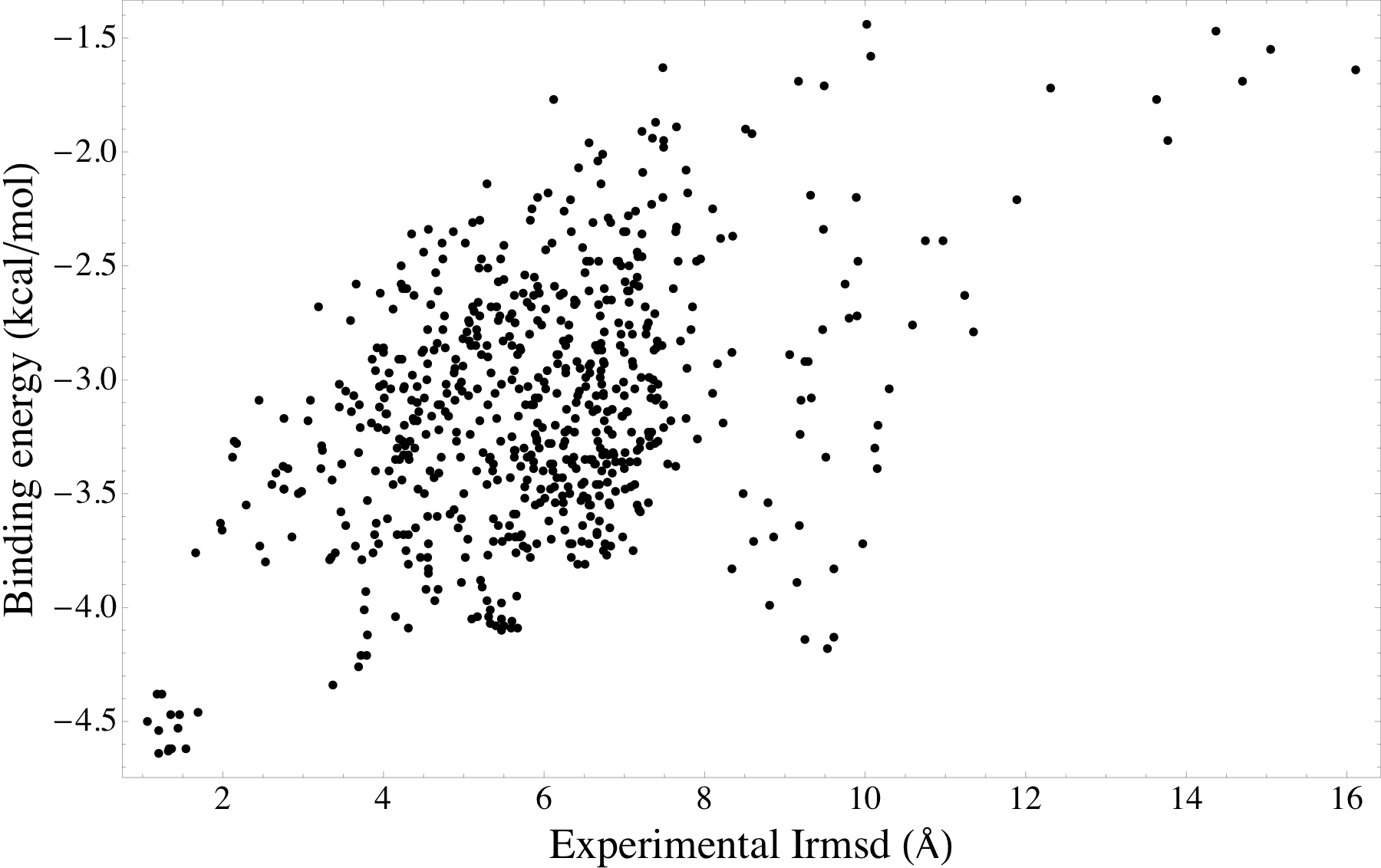
|
The clustering tree of the binding mode predictions between β2 adrenergic receptor and carazolol. The leaves represent docking complexes. The branches represent the structural distances between ligand conformations. Complexes were scored using the LMAPS module, from energetically unfavorable (blue) to favorable (crimson). The LMAPS module ensures that similar binding modes feature close scores. |
Energy landscape of the β2 adrenergic receptor - carazolol modeled complexes. Here, the modeled complexes with low energy binding modes are also similar to the experimental β2 adrenergic receptor - carazolol complex. |