Extraction d'information en génomique (Caderige)
Extraction d'information en génomique (Caderige)

Extraction d'information en génomique (Caderige)
Mots clef
extraction de connaissance, acquisition de connaissance, apprentissage automatique à partir de corpus,
traitement automatique de la langue, génomique.
Cette activité de MIG s'inscrit dans le cadre du projet Caderige.Contexte biologique
Les programmes de génomique génèrent des masses de données qui sont de plus en plus systématiquement répertoriées dans des collections internationales en ligne, qu'elles soient génériques, ou spécialisées par espèce ou par fonction. Cette information concerne les séquences et les structures annotées, des résultats de prédictions ou d'expériences à grande échelle de séquençage, de cartographie, ou de génomique fonctionnelle. Par contre, les connaissances plus fines sur la régulation de l'expression des gènes, qui sont nécessaires à la compréhension des mécanismes cellulaires, ne sont décrites que sous la forme des travaux publiés dans la littérature scientifique. Dès lors, l'accès à cette information documentaire est un enjeu central dans la construction de modèles d'interactions entre gènes et protéines, car c'est par ce biais que les chercheurs peuvent valider leurs hypothèses, voire définir de nouveaux plans d'expérience. Dans ce cadre, si la recherche d'information à l'aide de mots-clefs offre des performances intéressantes en terme de rapidité de traitement, les résultats renvoyés ne sont pas directement exploitables et nécessitent un important travail d'analyse des documents sélectionnés pour extraire l'information pertinente.Objectifs informatiques
L'objectif est de concevoir des méthodes d'automatisation de cette extraction. Le résultat de l'extraction formalisé dans un formulaire pourra être stocké dans une base de données ou de connaissances.
- Exemple
Dans le domaine de la génomique, lorsque, suite à un processus de recherche d'information sur le gène dacB, un document contenant le fragment de texte suivant est retourné : "... en ce qui concerne le gène dacB, on observe une bonne ressemblance entre son promoteur de transcription et la séquence reconnue par la sous-unité sigma E de l'ARN polymérase ..." le système devra pouvoir retourner automatiquement le formulaire ci-contre.
- Analyse linguistique et conceptuelle
L'acquisition de ce type de réponse nécessite la mise en oeuvre de méthodes d'analyse linguistique et conceptuelle permettant d'interpréter les documents et de construire dynamiquement les réponses appropriées. Ainsi, dans l'exemple précédent, le système doit prendre en compte une connaissance du domaine exprimant que s'il y a une bonne ressemblance entre le promoteur d'un gène et la séquence reconnue par un facteur sigma (ce qui est dit dans le fragment de document), alors ce facteur sigma contrôle très probablement l'expression de ce gène (ce qui est un type d'information précédemment défini comme pertinent dans le cadre de l'étude d'une interaction gène-protéine).
- Recours aux ontologies
Concrètement, le problème d'extraction de connaissances structurées se ramène donc à un problème d'appariement entre l'ensemble des représentations des informations à extraire définies par l'utilisateur, et les représentations des fragments de documents étudiés. Pour ce faire, ces différentes représentations doivent rendre compte des modèles recherchés par les utilisateurs dans les documents, ce qui oblige à effectuer des descriptions au niveau conceptuel. Ce type d'analyse complexe nécessite l'utilisation de thésaurii et d'ontologies spécifiques au domaine d'application. Malheureusement, ces ressources sont, en règle générale, non seulement inexistantes, mais également fort longues à acquérir " à la main ".
- Apprentissage automatique de connaissances à partir de texte
Du point de vue informatique, le point central concerne donc le développement de nouvelles méthodes automatiques et d'assistances à l'acquisition de telles ressources qui permettront de faire correspondre les modèles répertoriés des utilisateurs à leurs multiples manifestations langagières, de manière à obtenir des représentations conceptuelles canoniques des fragments de texte étudiés.
Ces différentes techniques informatiques seront validées en analysant les notices bibliographiques de MedLine concernant l'étude des interactions moléculaires. À terme, ce projet permettra donc une amélioration notable de l'exploitation qui est faite des ressources documentaires dans le domaine de la recherche d'interaction mais pas exclusivement dans celui-ci. En effet, dans la mesure où les méthodes informatiques qui seront développées sont basées, d'une part, sur l'apprentissage automatique de ressources lexicales et conceptuelles et d'autre part, sur des méthodes génériques d'analyse de la langue, elles seront transposables aux autres disciplines présentant le même type de phénomènes linguistiques. Ainsi nos méthodes seront exploitables dans l'ensemble du contexte de l'extraction de connaissance à partir de documentations scientifique et technique.
En biologie moléculaire, l'état de l'art est très en-deçà de ce qui est proposé ici. Les deux approches majoritaires consistent à construire des patrons manuellement [Blaschke et al., 99], [Thomas et al., 2000], [Ono et al., 2001], et le taux de rappel (de couverture), se situe par exemple entre 0 % et 30 % pour le problème d'identification des interactions géniques, ou à construire les patrons de faible précision sur des bases de co-occurrence statistique [Craven, 99], [Stapley & Benoit, 2000], [Pillet, 2000].Phases du projet
RéférencesBlaschke C., Andrade M. A., Ouzounis C. and Valencia A., "Automatic Extraction of biological information from scientific text: protein-protein interactions", in Proceedings of 7th International Conference on Intelligent Systems in Molecular Biology, (ISMB'99), p. 60-67, Heidelberg, Germany, AAAI Press, 1999.
Craven M. and Kumlien J., "Constructing Biological Knowledge Bases by Extracting Information from Text Sources", in Proceedings of the 7th International Conference on Intelligent Systems in Molecular Biology (ISMB-99), pp. 77-86, Heidelberg, Germany, AAAI Press, 1999.
Ono T., Hishigaki H., Tanigami A. and Takagi T., "Automated extraction of information on protein-protein interactions from the biological literature", in Bioinformatics vol 17, n° 2, pp. 155-161, Oxford Press, 2001.
Pereira F., Tishby N. and Lee L., "Distributional clustering of English words", in Proceedings of the Conference of the Association of Computational Linguistics (ACL'93), pp. 183-190, 1993.
Pillet V., Méthodologie d'extraction automatique d'information à partir de la littérature scientifique en vue d'alimenter un nouveau système d'information, thèse de l'Université de droit, d'économie et des sciences d'Aix-Marseille, 2000.
Stapley B. J. and Benoit G., "Bibliometrics: Information Retrieval and Visualization from Co-occurrence of Gene Names in MedLine Abstracts", In Proceedings of the Pacific Symposium on Biocomputing (PSB'2000), vol. 5, pp. 529-540, Honolulu, 2000.
Thomas, J., Milward, D., Ouzounis C., Pulman S. and Caroll M., "Automatic Extraction of Protein Interactions from Scientific Abstracts", in Proceedings of the Pacific Symposium on Biocomputing (PSB'2000), vol.5, p. 502-513, Honolulu, 2000.
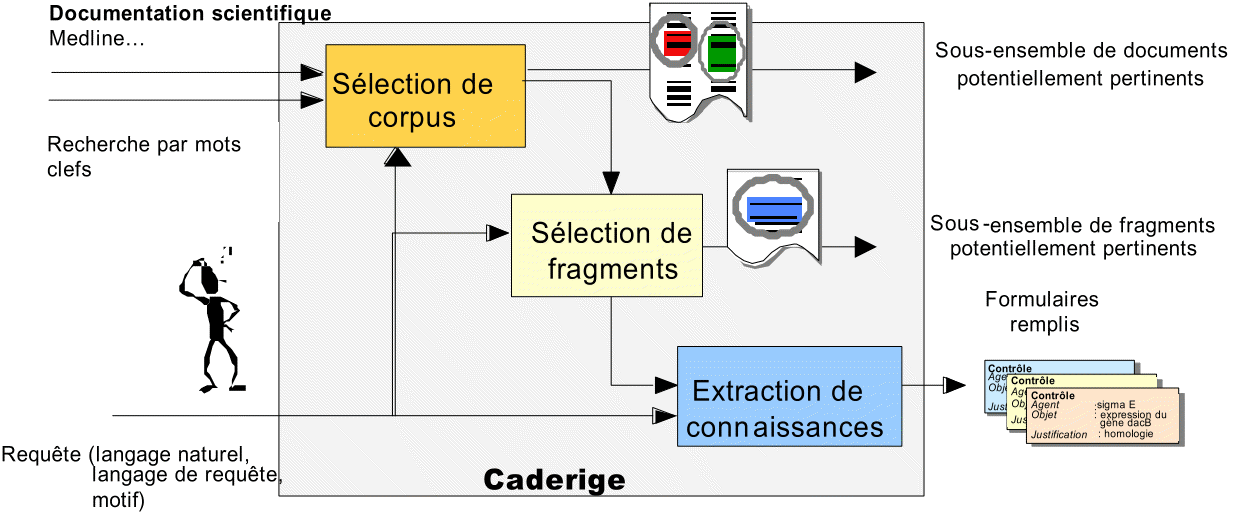
Nous abordons trois méthodes différentes d'exploitation d'une documentation scientifique : il s'agit de sélectionner les textes pertinents, de sélectionner les fragments les plus intéressants et enfin d'extraire de la connaissance véhiculée par les textes pour en donner une représentation formelle.
Ces trois méthodes sont complémentaires et reposent sur des techniques apparentées. Le processus devra se dérouler en trois étapes. Des traitements de complexité croissante sont effectués à chaque étape, mais les traitements intermédiaires apportent déjà une aide appréciable en matière d'exploration documentaire :
- La première étape " Sélection de corpus " consiste à constituer un corpus en sélectionnant au sein de la base documentaire les documents effectivement pertinents au regard de la requête formulée par l'utilisateur. Ces corpus sont directement exploitables par les utilisateurs comme source d'information et ils constituent le corpus approprié sur lequel devra porter l'extraction automatique d'information.
Cette sélection repose sur un classifieur de documents qui dépend lui-même des types de documents considérés et des critères de pertinence de l'utilisateur.
- La deuxième étape " Sélection de fragments " permet d'identifier dans les documents du corpus sélectionné à l'étape précédente, les portions de texte qui concernent effectivement le problème applicatif posé. Ils sont généralement très dispersés, par exemple les phrases sur les interactions géniques forment 3 % des résumés sur le sujet. Cette étape de filtrage est donc essentielle, que ce soit pour présenter directement les fragments sélectionnés à l'utilisateur sous la forme de texte surligné et comme une aide à la lecture les documents, ou pour aborder le traitement lourd d'extraction de connaissance proprement dit.
Dans le domaine de la génomique fonctionnelle, pour identifier les interactions fonctionnelles entre les objets biologiques, problème clef de la génomique fonctionnelle, il est important de savoir repérer dans les résumés d'articles scientifiques de Medline des fragments tels que celui de la figure 2 (colonne de gauche).
De la même manière que pour la sélection de corpus, cette sélection de fragments repose sur un classifieur de phrases.
- La troisième étape " Extraction de connaissance " permet d'extraire et de donner une représentation formelle de l'information pertinente. La figure 2 montre quel type de connaissance le module d'extraction vise à extraire. Cette connaissance est représentée sous la forme de formulaires (colonne de droite sur la figure 2). Du fragment 2, on retient ainsi l'information que HIPK2 appartient à la famille des kinases sérine/threonine et que HIPK2 interagit positivement (activation) sur le gène p53.
De manière classique, cette étape d'extraction repose sur un moteur d'extraction qui confronte au texte un ensemble de règles, de patrons ou schémas de phrases (extraction patterns) qui modélisent les informations recherchées (exemple, figure 2).
Fragment 1 Fonctionnal role Source : protein HIPK2
Target : apoptosis"Furthermore, HIPK2 and p53 cooperate in the activation of p53-dependant transcription and apoptotic pathways." Fonctionnal role Source : protein p53
Target : apoptosis
Fragment 2 Membership Element : protein kinase-2
Family : serine /threonine kinases
"Here we demonstrate that homeodomain-interacting protein kinase-2, a member of a novel family of nuclear serine/threonine kinases, activates p53 by directly phosphorylating it at Ser 46." Interaction Source : protein kinase-2
Target : p53
Type : PositiveFigure 2. Des fragments de texte, aux formulaires remplis.
Claire Nédellec |
Philippe Bessières |