Contrôle qualité des données issues des technologies de séquençage avec FastQC et MultiQC
Outils présentés
- FastQC v0.11.5
- multiQC v1.0
Principe
Évaluer la qualité des données issues d'un séquençage haut-débit est la première étape à effectuer avant de se lancer dans des analyses bioinformatiques. Cela permet de déterminer si les données qui vous sont fournies sont conformes à ce que vous aviez demandé au prestataire de séquençage. Généralement, cette étape est effectuée par le prestataire, mais mieux vaut vérifier par vous-même pour éviter les mauvaises surprises et pour commencer à vous familiariser avec vos données. Il est important de déterminer, entre autres :
- le nombre de lectures (reads)
- la qualité des lectures
- la longueur des lectures
- une éventuelle contamination
- la présence suspecte d'adaptateurs
- ...
Les données vont sont fournies généralement au format FASTQ. Si ce n'est pas le cas, il existe des outils de conversion pour obtenir des fichiers FASTQ.
Le format FASTQ
Le format FASTQ est le format de référence des reads bruts issus d'un séquençage haut-débit. Il s'agit d'un fichier texte qui se compose d'informations relatives à chaque séquence. Ces informations sont regroupées sur 4 lignes.
Exemple d'une séquence de 15 nucléotides au format FASTQ :
@M02944:130:000000000-ARV3J:1:1102:11673:1783 1:N:0:CACCGG
CTTGGTCATTTAGAG
+
***<<*AEF???***
- Ligne 1 : le nom de la lecture et des métadonnées propres au séquençage. Elle commence toujours par le caractère @
- Ligne 2 : la séquence
- Ligne 3 : un séparateur (caractère +) pouvant être suivi des mêmes informations que sur la ligne 1
- Ligne 4 : les valeurs de qualité de chaque base de la lecture
La qualité de chaque base est encodée de façon à n'être représentée qu'avec un caractère, de façon à limiter la taille du fichier et faciliter sa compression.
Cette ligne n'est pas interprétable à l'oeil mais les logiciels sauront décoder l'information. Il existe différentes plages de qualité qui sont utilisées en fonction du type de séquençage effectué.

Le score de qualité est relié de façon logarithmique à la probabilité d'erreur. Cette probabilité d'erreur est calculée lors de l'identification d'une base et correspond à la probabilité qu'il s'agisse d'une mauvaise identification.
Concrètement, un score de 20 signifie qu'il existe une chance sur 100 pour que la base soit fausse. Un score de 30 à une chance sur 1000.
En fonction du type de données que vous allez traiter, il est possible que vous ayez envie/besoin de supprimer de vos reads des bases de mauvaise qualité (présentes souvent aux extrémités 3' des reads). Cette étape s'appelle le trimming des reads et repose en partie sur l'information que vous aurez obtenue de l'analyse qualité. Un tutoriel pour les outils de trimming sera bientôt à disposition.
Pour plus d'informations sur le format FASTQ, rendez-vous ici
FastQC
Il existe de nombreux outils bioinformatiques permettant d'analyser la qualité des données de séquençage (voir Références).
Pour un usage généraliste, FastQC est particulièrement adapté. Il permet une visualisation graphique des différentes métriques d'intérêt avec un code couleur.
Voici les informations que l'on peut obtenir en utilisant FastQC :
Per Base Sequence Quality
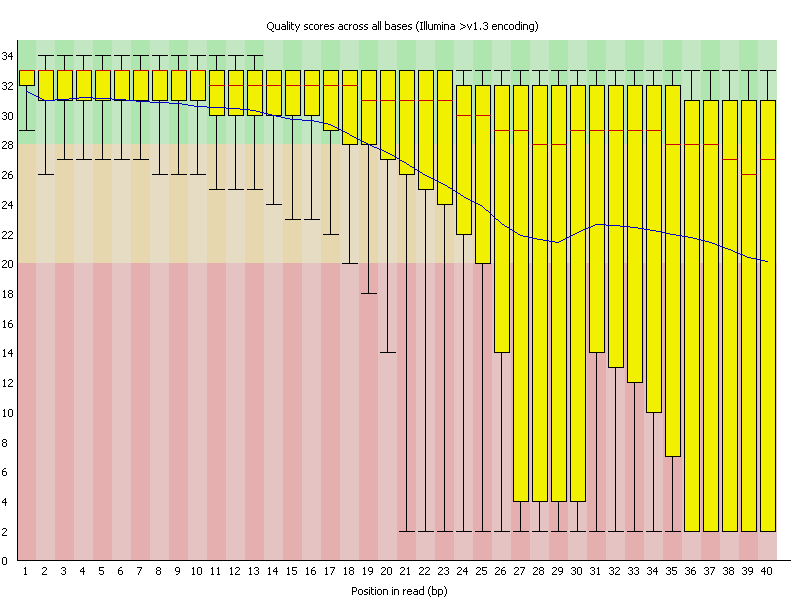
Ce graphique représente la qualité (score Phred, en ordonnée) de chaque base (en abscisse) pour tous les reads de votre jeu de données. À chaque position du read, la qualité de tous les reads est représentée sous la forme d'un boxplot. La médiane est en rouge. Le code couleur vous indique les scores de très bonne qualité en vert, bonne qualité en orange et mauvaise en rouge. Généralement, la qualité baisse en fin de reads.
C'est ce graphique qui va vous indiquer si il faut trimmer vos reads ou non.
Per Sequence Quality Scores
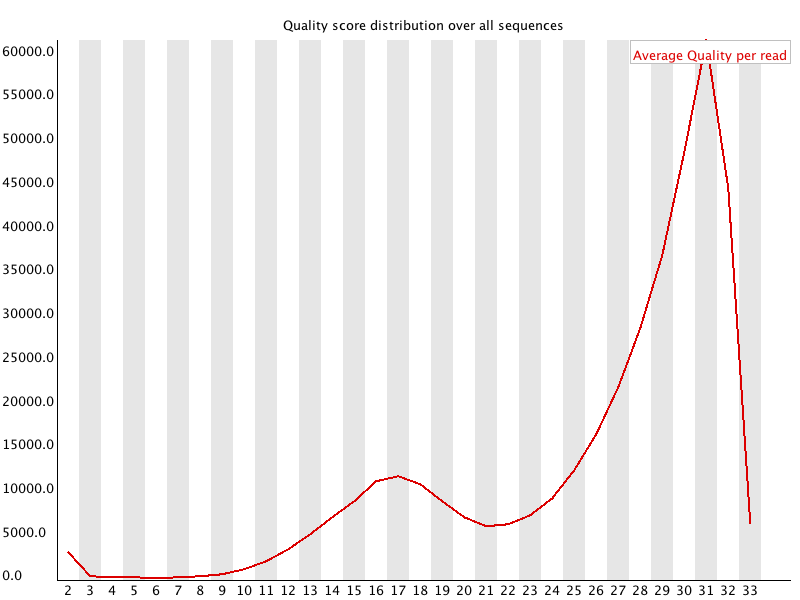
Ce graphique représente le nombre de reads pour chaque score de qualité. Il est très informatif s'il y a eu un problème lors du séquençage.
Per Base Sequence Content
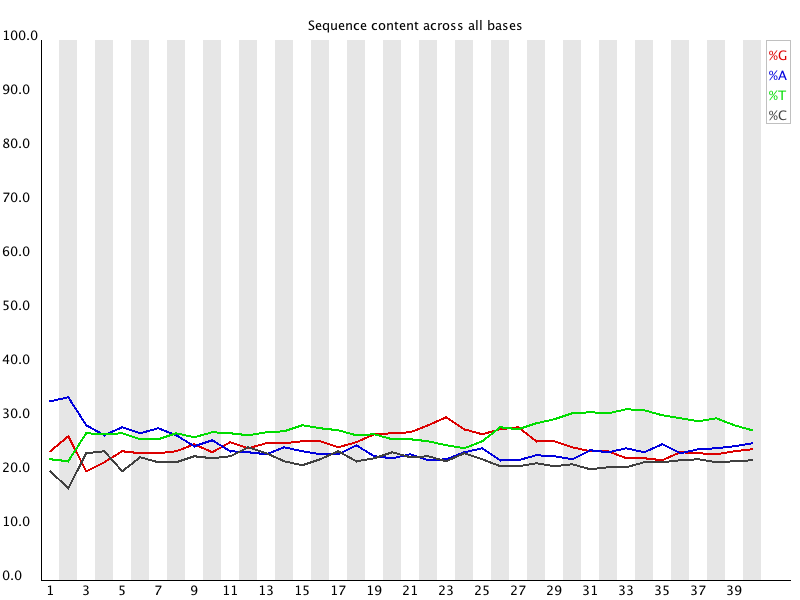
Ce graphique représente le % de chaque base à chaque position du read.
Attention au type de données que vous manipulez ! FastQC étant à l'origine conçu pour des données génomiques, certaines métriques peuvent être indiquées comme mauvaises (croix rouge) alors que ce n'est pas réellement le cas.
Si vous analysez des données transcriptomiques, la composition en bases peut être fortement impactée par des séquences sur-représentées. La présence d'adaptateurs perturbe également la composition.
Soyez donc vigilent lors de l'interprétation.
Voici un exemple tout à fait normal de données amplicons, bien qu'annoté avec une croix rouge :

Sequence Length Distribution
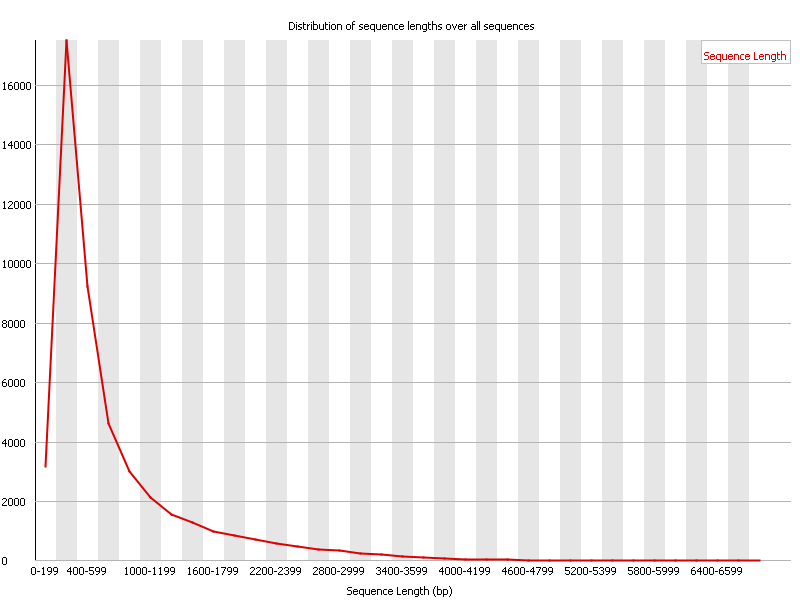
Ce graphique représente la distribution des longueurs de vos séquences. Il est inutile si vous vous attendez à avoir toutes les séquences de la même taille comme lors d'un séquençage Illumina. Il peut être très intéressant dans le cas d'une autre technologie (PacBio ou autre).
Et encore...
Voici un rapport FastQC complet. C'est celui que l'on vous propose de générer dans le tutoriel :
Sur le site de
FastQC, vous trouverez le détail de l'ensemble des graphiques proposés, des exemples de résultats pour des données issues de différentes technologies de séquençage (Illumina, 454, PacBio...), et des exemples de bons et de mauvais runs.
MultiQC
MultiQC quant à lui permet de regrouper les informations provenant de multiples rapports FastQC. Il est très utile dès qu'on dépasse quelques échantillons à traiter. Il regroupe au sein d'une même page toutes les informations provenant de multiples rapports et permet une comparaison facilitée des échantillons. Il est alors aisé de repérer un ou plusieurs échantillons différents des autres.
Voici le rapport que l'on vous propose de générer dans le tutoriel :
Autres fonctionnalités de MultiQC
MultiQC va plus loin que résumer des sorties de FastQC. Il est capable de parser les logs générés par
cutadapt,
jellyfish,
sortMeRNA,
bowtie2,
samtools,
GATK,
TopHat, et des dizaines d'autres outils bioinformatiques.
Pour illustration, voici un exemple d'un rapport regroupant une analyse FastQC, STAR, Cutadapt et featureCounts.
Analyse en temps réel
Connexion au serveur migale
Dans un premier temps, connectez-vous sur le serveur migale avec votre identifiant et votre mot de passe. Si vous ne possédez pas de compte, voici le formulaire dédié. Vous trouverez la procédure pour vous connecter à partir de Windows ici.
Il faut que l'option -X ou -Y de ssh ait été rentrée au moment de votre connection au serveur migale, ceci afin d'autoriser l'affichage graphique.
Préparation de votre espace de travail
Créez un répertoire de travail dans votre home, déplacez-vous à l'intérieur et faites un lien symbolique des données de séquençage.
mkdir ~/TUTO_FASTQC
cd ~/TUTO_FASTQC
ln -s /usr/local/genome/tutorials/QualityControl/Single/*.gz .
Le lien symbolique comme son nom l'indique permet de pointer vers un fichier sans le dupliquer. C'est essentiel pour économiser de l'espace disque. Attention, toute modification du fichier d'origine sera répercutée sur le lien. Si le fichier d'origine est déplacé ou supprimé, le lien est cassé et vous n'y avez plus accès.
Pour connaître la version de Fastqc disponible, il suffit de taper :
fastqc --version
Options principales :
- -t INT : pour paralléliser le traitement
- -c FILE : pour fournir une liste de séquences à considérer comme contaminants
- -a FILE : pour fournir une liste de séquences à considérer comme adaptateurs
- -l FILE : pour reconfigurer les seuils limites (warning/error)
Pour obtenir la liste complète des options, il suffit de taper :
fastqc -h
Lancement de FastqC sur un fichier
Aucun traitement ne doit être lancé sur le serveur migale ! Il va falloir déporter le traitement sur un noeud du cluster de calcul. Ici nous lancerons le traitement avec la commande qsub mais il est également possible de se connecter directement sur un noeud. Plus d'informations ici.
La commande qsub permet de soumettre un job sur un noeud de calcul. Plus d'infos sur la commande qsub
ici.
qsub -cwd -V -N FASTQC -b y "fastqc SRR1145846_S1_L001_R1_001.fastq.gz"
Utilisez la commande qstat pour vérifier l'état de votre job sur le cluster. Plus d'infos sur la commande qstat
ici.
Exploration des fichiers générés par FastQC
Fichiers générés par fastqc:
Tapez la commande ls pour lister les fichiers présents dans votre répertoire.
ls -l
- SRR1145846_S1_L001_R1_001_fastqc.html : le rapport HTML produit par FastQC
- SRR1145846_S1_L001_R1_001_fastqc.zip : une archive contenant toutes les images visibles dans le rapport HTML.
- Les fichiers FASTQC.e******* et FASTQC.o******* ne sont pas générés par l'outil mais par SGE (le système de gestion du cluster de calcul). Dans le fichier .o se trouvent généralement les logs écrits par l'outil et dans le fichier .e les erreurs éventuelles.
Visualisation du rapport FastQC
Pour visualiser un fichier .html sur migale, il faut lancer Firefox :
firefox SRR1145846_S1_L001_R1_001_fastqc.html
Lancement de FastqC sur tous les fichiers d'un répertoire
Imaginons que vous ayez 600 échantillons à analyser. Vous ne pouvez pas vous permettre de lancer 600 fois la commande précédente. Il va donc falloir automatiser le traitement.
De plus, il n'est pas envisageable d'ouvrir les 600 rapports pour vérifier la qualité de vos données. L'outil MultiQC a été conçu pour résoudre ce problème.
Nettoyage de l'espace de travail
Dans un premier temps, effacez tout le contenu du répertoire TUTO_FASTQC avec la commande rm. Ensuite, créez des liens symboliques vers tous les fichiers se terminant par .gz du répertoire /usr/local/genome/tutorials/QualityControl/Multiple/.
rm -Rf ~/TUTO_FASTQC/*
ls
ln -s /usr/local/genome/tutorials/QualityControl/Multiple/*.gz .
ls
Vous devriez avoir dans votre répertoire tous ces fichiers, 78 au total :
CD110CE_R1.fastq.gz CD116CE_R1.fastq.gz CD210CE_R1.fastq.gz CD216CE_R1.fastq.gz CD310CE_R1.fastq.gz CD316CE_R1.fastq.gz CDM13_R1.fastq.gz
CD110CE_R2.fastq.gz CD116CE_R2.fastq.gz CD210CE_R2.fastq.gz CD216CE_R2.fastq.gz CD310CE_R2.fastq.gz CD316CE_R2.fastq.gz CDM13_R2.fastq.gz
CD111CE_R1.fastq.gz CD15CE_R1.fastq.gz CD211CE_R1.fastq.gz CD25CE_R1.fastq.gz CD311CE_R1.fastq.gz CD35CE_R1.fastq.gz CDM4_R1.fastq.gz
CD111CE_R2.fastq.gz CD15CE_R2.fastq.gz CD211CE_R2.fastq.gz CD25CE_R2.fastq.gz CD311CE_R2.fastq.gz CD35CE_R2.fastq.gz CDM4_R2.fastq.gz
CD112CE_R1.fastq.gz CD16CE_R1.fastq.gz CD212CE_R1.fastq.gz CD26CE_R1.fastq.gz CD312CE_R1.fastq.gz CD36CE_R1.fastq.gz CDP13_R1.fastq.gz
CD112CE_R2.fastq.gz CD16CE_R2.fastq.gz CD212CE_R2.fastq.gz CD26CE_R2.fastq.gz CD312CE_R2.fastq.gz CD36CE_R2.fastq.gz CDP13_R2.fastq.gz
CD113CE_R1.fastq.gz CD17CE_R1.fastq.gz CD213CE_R1.fastq.gz CD27CE_R1.fastq.gz CD313CE_R1.fastq.gz CD37CE_R1.fastq.gz
CD113CE_R2.fastq.gz CD17CE_R2.fastq.gz CD213CE_R2.fastq.gz CD27CE_R2.fastq.gz CD313CE_R2.fastq.gz CD37CE_R2.fastq.gz
CD114CE_R1.fastq.gz CD18CE_R1.fastq.gz CD214CE_R1.fastq.gz CD28CE_R1.fastq.gz CD314CE_R1.fastq.gz CD38CE_R1.fastq.gz
CD114CE_R2.fastq.gz CD18CE_R2.fastq.gz CD214CE_R2.fastq.gz CD28CE_R2.fastq.gz CD314CE_R2.fastq.gz CD38CE_R2.fastq.gz
CD115CE_R1.fastq.gz CD19CE_R1.fastq.gz CD215CE_R1.fastq.gz CD29CE_R1.fastq.gz CD315CE_R1.fastq.gz CD39CE_R1.fastq.gz
CD115CE_R2.fastq.gz CD19CE_R2.fastq.gz CD215CE_R2.fastq.gz CD29CE_R2.fastq.gz CD315CE_R2.fastq.gz CD39CE_R2.fastq.gz
Automatisation
Créez dans un premier temps un répertoire FASTQC et un répertoire LOGS avec la commande mkdir.
Ensuite, la commande écrite en bash permet d'écrire dans le fichier fastqc.sh la commande qsub pour chaque fichier .gz présent dans le répertoire.
Vérifiez que le fichier est bien formé avec la commande cat.
Vérifiez que vous avez bien 78 lignes dans le fichier fastqc.sh avec la commande wc -l.
Enfin lancez vos commandes avec la commande sh.
mkdir LOGS
mkdir FASTQC
for i in *.gz ; do echo "qsub -cwd -V -N FASTQC -o LOGS/ -e LOGS/ -b y 'fastqc $i -o FASTQC'" >> fastqc.sh ; done
cat fastqc.sh
wc -l fastqc.sh
sh fastqc.sh
Le répertoire LOGS a été créé pour y déposer tous les fichiers .e et .o générés par chaque commande qsub. Le répertoire FASTQC est la destination des rapports générés par chaque commande.
Difficile d'ouvrir et de comparer les 78 rapports produits. Nous allons donc utiliser MultiQC, un outil qui va nous permettre d’avoir une vue synthétique sur l’ensemble des résultats.
Lancement de MultiQC
MultiQC va permettre de mutualiser toutes les informations contenues dans les 78 rapports produits. L'outil prend en entrée un répertoire dans lequel sont contenus les logs produits par FastQC.
Tout d'abord, créez un répertoire MULTIQC puis lancez la ligne de commande permettant de lancer multiqc sur le cluster de calcul.
Attention, multiqc fait partie des logiciels python qui nécessitent de charger leur propre environnement avant d'être accessibles et lancés. Plus d'informations
ici.
mkdir MULTIQC
qsub -cwd -V -N FASTQC -b y "source multiqc.env ; multiqc -d FASTQC/ -i QualityControl -o MULTIQC/ -n quality_control ; deactivate"
Options de multiqc :
- -i : nom à donner à l'analyse (sera utilisé dans le rapport)
- -o : répertoire où écrire les sorties de multiQC
- -n : nom du fichier de sortie, qui sera suffixé par .html
- --ignore : répertoires à ignorer (expressions régulières possibles)
- --file-list : fichier contenant les fichiers à scanner
- ...
Il existe de plus des options pour choisir quel(s) modules exécuter, quel template utiliser... La documentation complète est disponible
ici.
Visualisation du rapport MultiQC
Utilisez firefox pour visualiser le rapport HTML.
firefox MULTIQC/quality_control.html &
Vous pouvez maintenant visualiser et comparer la qualité et les informations concernant vos jeux de données très facilement. De plus, vous avez le menu Toolbox sur la droite de la page qui vous permet de choisir les échantillons à visualiser, d'exporter les images...
Analyse en temps réel
Connexion au portail Galaxy
Ouvrez un navigateur web et entrez l'url migale.jouy.inra.fr/galaxy/. Entrez vos login et mots de passe (identiques à ceux utilisés pour se connecter au serveur migale).
Vous trouverez ici les informations nécessaires à l'utilisation du portail Galaxy mis à votre disposition par la plateforme migale.
Préparation de votre espace de travail
Création d'un nouvel historique
Créez un nouvel historique que vous nommerez QUALITY_CONTROL
Import des fichers de travail
Les fichiers que vous allez manipuler sont stockés à l'intérieur d'une DataLibrary. Pour importer les fichiers :
- Cliquez sur Shared Data
- Cliquez sur Data Libraries
- Cliquez sur Tutorial Quality Control
- Cochez la case à côté de FASTQ files
- Cliquez sur le bouton to History
- Cliquez sur le bouton Import
- Cliquez sur Analyze Data (menu du haut) pour revenir à votre historique
Vous avez à présent 11 datasets dans votre historique. Un fichier est au format fastq.gz : il est compressé. Vous ne pourrez pas le visualiser. Les autres sont au format FASTQ.

Lancement de FastqC sur un fichier
Cliquez sur l'outil FastQC Read Quality reports, qui est rangé dans la section NGS: QC and manipulation.
Remplissez le formulaire puis cliquez sur Execute
- Short read data from your current history : sélectionnez le fichier FASTQ SRR1145846_S1_L001_R1_001.fastq.gz de l'historique.
- Contaminant list : Nothing selected.
- Submodule and Limit specifing file : Nothing selected.

L'outil génère deux fichiers :
- Un dataset Webpage au format html qui contient le rapport FastqC qui est généré.
- Un dataset RawData au format txt qui contient un tas d'informations. Ce sont les valeurs numériques qui sont représentées sous forme de graphiques dans le rapport html.

Lancement de FastqC sur plusieurs fichiers
Pour lancer l'outil astQC Read Quality reports sur plusieurs fichiers, il n'est pas nécessaire de relancer l'outil autant de fois qu'il y a de fichiers. Une fonctionnalité très intéressante de Galaxy permet de lancer un outil sur plusieurs fichiers. Il suffit de sélectionner l'option Multiple Datasets et de sélectionner les fichiers sur lesquels vous voulez lancer l'outil.


Lancement de MultiQC
Cliquez sur l'outil multiqc aggregate results from bioinformatics analyses across many samples into a single report, qui est rangé dans la section NGS: QC and manipulation.
Remplissez le formulaire puis cliquez sur Execute
- Software name : choisissez FastQC (RawData file).
- Result file : sélectionnez les fichiers que vous voulez visualiser.


Le rapport est visualisable en cliquant sur l'icône en forme d'oeil du dataset dans lequel est écrit Webpage. L'autre dataset étant un fichier contenant les logs du process.
Outils bioinformatiques
FastQC
S. Andrews (2010), FastQC: a quality control tool for high throughput sequence data. Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc
MultiQC
Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016. doi:10.1093/bioinformatics/btw354
Fastqp
M. Shirley (2014), Simple FASTQ, SAM and BAM read quality assessment and plotting using Python. Available online at: https://github.com/mdshw5/fastqp
Prinseq
Schmieder R. (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics, 2011 Mar 15;27(6):863-4
RSeqC
Wang LG, Wang SQ, Li W: RSeQC: quality control of RNA-seq experiments. Bioinformatics 2012,28(16):2184–2185
RNASeqC
DeLuca,D.S.et al. (2012) RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics,28,1530–1532