Documentation
YeastIP is a dynamic database gathering verified hemiascomycetous yeasts sequences of the most widely used markers for phylogeny.
- LSU complete: complete sequence of the 26S ribosomal RNA gene (large subunit)
- LSU D1/D2: partial sequence of the 26S ribosomal gene comprising the D1/D2 region
- SSU complete: complete sequence of the 18S ribosomal RNA gene (small subunit)
- ITS ribosomal RNA region containing the intergenic region 1 (between 18S and 5.8S), the 5.8S ribosomal RNA gene and the intergenic region 2 (between 5.8S and 26S)
- mtSSU: mitochondrial small subunit ribosomal RNA gene
- RPB1: RNA polymerase II largest subunit gene
- RBP2: partial sequence of the RNA polymerase II second largest subunit gene
- TEF1-alpha: partial sequence of the translation elongation factor 1-alpha gene
- ACT1: partial sequence of the exon2 of the ACT1 gene coding for actin
- mtCOX II: partial sequence of mitochondrial cytochrome C oxidase subunit 2 gene
- T: type strain
- NT: neotype
- ST: syntype
- HT: holotype
- LT: lectotype
- IT: isotype
- A: authentic strain
- N: not type
The name of genus and species registered as Current Name in the database is the most recent name, according to the most recent
taxonomic studies. Synonyms are listed in each sequence file, and are searchable via the Search by keyword tool (see 3.).
The search by listbox gives only the most recent name (see 1.b and 3.).
Each sequence file contains a clickable NCBI accession number (except for sequence extracted from complete genome sequence),
definition of the sequence (i.e. description in NCBI), name of the marker, strain status, sequence, number of Ns in the sequence,
length of the sequence, clickable PMID number, reference of the taxonomic study, current name, variety, clade, synonyms and
comments on the sequence.
Homepage indicates some statistical information, like the number of sequences, clades, genera and species currently in the database.
It also gives the distribution of sequences per marker.
The authentication tool allows the user to compare his own sequence to the YeastIP database using the Blast tool. The sequence of
interest must be pasted in the first frame, and, if necessary, could be given a title in the second frame (not required , by default
the title is Query). In the drop-down menu, a choice of databases is offered: whole database or only type strain sequences. The
Blast options are: blastn, -F F -I -J -T T -v 75 -W 28 -K 100 (see Blast options).
Blast results appear at the end of the page. The scrolling bar allows to consult the alignments. It is possible to retrieve the
sequences of the best last results as fasta format at the top of the page (Get Fasta sequences button). The number of sequences
to retrieve may be chosen by the user in the dedicated box.
The program also indicates in red the identification result (best Blast result), together with a list of the markers available for
this species in the database. The user must be careful with this information, because with some markers like D1/D2, more than one species
have the same sequence. For best use of the database, the user is adviced to examine alignments derived from the blast search.
To see the available markers for the species from the blast result, enter the number of species to display and click Marker
table'. This will open a page presenting table with all the selected species in line and the marker in column. Intersection will
give the presence (in green) or absence (in red) of the considered markers for each species in the database. This page also contains
a link to the phylogeny tool (see 4.).
YeastIP database allows the search and the retrieval of sequences for taxonomy and phylogeny with the Sequence search tool. It can
be questioned by:
o Keywords:
The Keywords box is not case sensitive but is sensitive to spelling. For example, the word cndida will
return no match, but cAndI will. Keywords interrogate the database in all fields: name, synonym, markers, collection numbers
(most of them are CBS number) etc...
o Genus or species name:
For a specific search by species name or markers, it is recommanded to use the second part of the
search tool. For a search by name, select the genus first, then the associated species name. The latter will appear in a list
box. If the species of interest does not appear, either it is not in the database, or it is a synonym of the name registered in the
database. To choose between, these two possibilities, the user should questioned the database by keyword.
o Clade:
The database also provides all registered strains in clades (see Phylogeny of yeast clade). Markers may be associated to this search,
but it is better to avoid a search combining clade and genus: if the selected genus does not belong to the selected clade, the
search will fail.
Markers may be chosen either as a unique search field (for example, display all D1/D2 present in the database), or combined
with a clade or genus/species search (for example, all the Candida D1/D2, or D1/D2 + ITS of Saccharomyces cerevisiae...).
Finally, the search may be refined by selecting only type strains with the checkbox (check by default).
The result will be displayed in a table containing the accession number, the status of the strain (type, neotype... see 1.b.),
the current species name to which the strain belongs, the collection number of the strain, the name of the marker, the length of
the sequence and the most recent synonym of the species cited in NCBI.
Sequence file may be opened by clicking the accession number to visualize information as described in 1.d. Checkboxes allow to
select sequences to be retrieved in fasta format, or to select the strain to be displayed in the marker table (see 4.a.). The display
of a strain in the marker table can simply be done by checking only one sequence of this strain.
The number of retrievable sequences is voluntary limited to 500. If your search contains more than 500 sequences, please select
more criteria to refine the search. For special demands, please contact us.
The phylogeny tool allows the selection of markers to use for concatenation, the addition of user sequences and the display of a
phylogenic tree.
The marker table may be reached in two ways: by a blast authentication, or by a search in the 'Sequence search' tool.
It displays all the current markers of the selected strain in the database. In regard of each column and row, the check boxes
allow selection of strains and markers for concatenation. At least one strain and one marker must be selected to pursue to the
concatenation tool. Be careful to select only markers which are present in all the selected strains. Then go to the Concatenation
button to obtain your selected sequences concatenated by species in the same order for all markers in Fasta format.
The concatenation file will open in another page. The complete sequence file can be retrieved in a Fasta format zipped file. Users who want to
add their own sequences to the selection have to enter
the number of strains that they want to add in the box and click 'Add'. This action will open another window with predeterminated boxes
whose number reflects the number of markers previously selected and the number of strains to add.
On the concatenation page, the 'Phylogeny.fr' button opens to the Phylogeny.fr website in a new tab. The concatenation file will
be automatically loaded and the 'One click' analysis will be launched. The tree will be displayed on the Phylogeny.fr website (Dereeper
A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.-F., Guindon S., Lefort V., Lescot M., Claverie J.-M., Gascuel O.
Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Research. 2008 Jul 1; 36 (Web Server Issue):W465-9. Epub
2008 Apr 19.).
Back to top
Marker choices
DNA markers do not provide the same information in taxonomy and phylogeny. While D1/D2 is the most widely used marker to describe
species, it is not very reliable for phylogeny because of its small size. Other markers are good performers in phylogenetic analysis,
but they are not available for a large number of species. Markers typically used for barcoding (ITS1-5.8S-ITS2, mtCOX II
) are not
reliable markers for phylogeny: for instance the size of ITS1-5.8S-ITS2 can be very variable, and mitochondrial markers such as
mtCOX II may be misleading because of the peculiar inheritance and the mode of evolution of mitochondrial DNA. Concatenation of
coding sequences like ACT1, RPB1, RPB2 or TEF1-alpha provide good robust phylogeny.
Back to top
Primers
Most of the oligonucleotide primers used to amplify DNA and generate sequences present in the database can be found in the work
by:
Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics.Taylor et al., 1990, PCR
Protocols: a Guide to Methods and Applications, (ed. M.A. Innis, D.H. Gelfand, J. Sninsky, T.J. White), pp. 315322. Academic press, San
Diego).
Identification and phylogeny of ascomycetous yeasts from analysis of nuclear large subunit (26S) ribosomal DNA partial sequences.
Kurtzman C.P. and Robnett C.J., 1998, Antonie van Leeuwenhoek , 73:331-371.
Partial sequence analysis of the actin gene and its potential for studying the phylogeny of Candida species and their teleomorphs.
Daniel H.M., Sorell T.C. and Meyer W., 2001, Int. J. Syst. Evol. Microbiol., 51:1593-1606.
Phylogenetic relationships among yeasts of the 'Saccharomyces complex' determined from multigene sequence analysis.
Kurtzman C.P. and Robnett C.J., 2003, FEMS Yeast Res., 3:417-432.
Phylogenetic circumscription of Saccharomyces, Kluyveromyces and other members of the Saccharomycetaceae, and the proposal of
the new genera Lachancea, Nakaseomyces, Naumovia, Vanderwaltozyma and Zygotorulaspora. Kurztman C.P., 2003, FEMS Yeast
Res., 4:233-245.
Evaluation of ribosomal RNA and actin gene sequences for the identification of ascomycetous yeasts. Daniel H.M. and Meyer W., 2003,
Int. J. Food Microbiol., 86:71-78.
Multigene phylogenetic analysis of Trichomonascus, Wickerhamiella and Zygoascus yeast clade, and the proposal
of Sugiyamaella gen. nov. and fourteen new species combinations. Kurtzman C.P. and Robnett C.J., 2007, FEMS Yeast Res., 7:141-154.
Re-examining the phylogeny of clinically relevant Candida species and allied genera based on multigene analysis. Tsui C.K.M.,
Daniel H.M., Robert V. and Meyer W., 2008, FEMS Yeast Res., 8:651-659.
Phylogeny and evolution of medical species of Candida and related taxa: a multigenic analysis.Diezman et al., 200), J. Clin.
Microbiol. 42: 5624-5635.
Phylogenetic relationships among species of Pichia, Issatchenkia and Williopsis
determined from multigene sequence analysis, and the proposal
of Barnettozyma gen. nov., Lindnera gen. nov. and Wickerhamomyces gen. nov.Kurtzman C.P. et al., 2008, FEMS Yeast Res. 8: 939-954.
Back to top
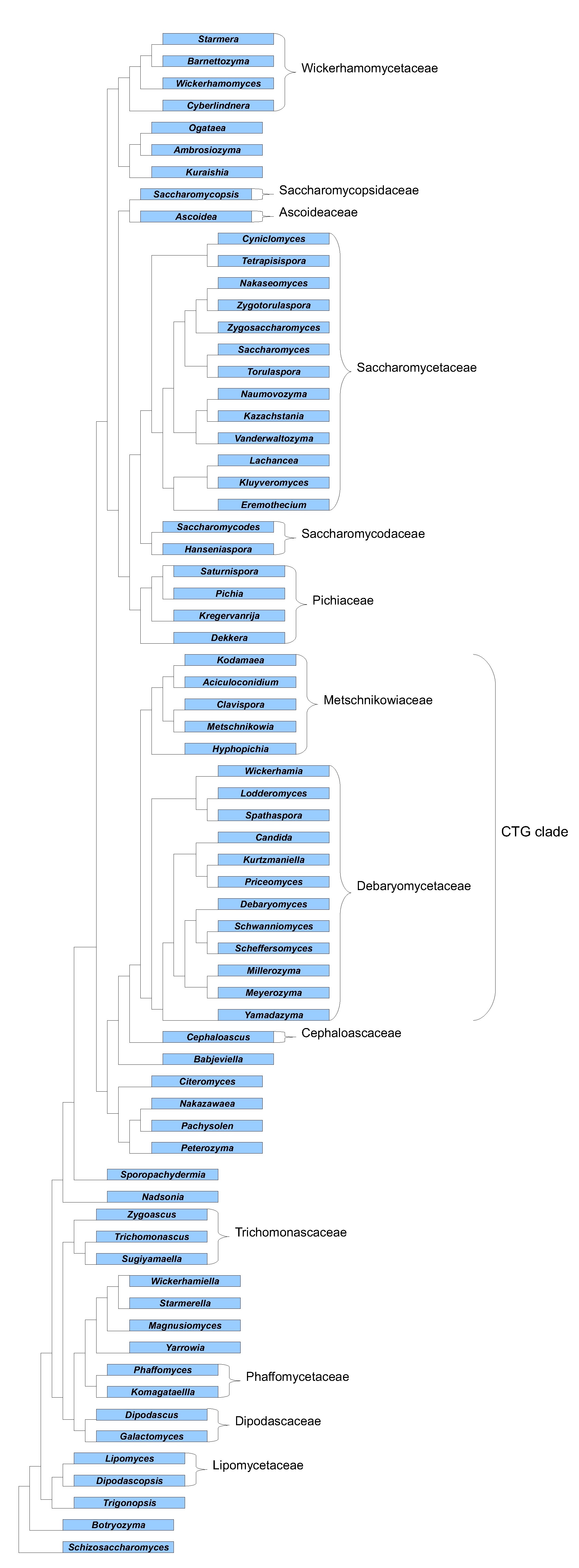
Phylogeny of yeast clades
The genus Candida contains species that have no sexual state. The Candida genus is not monophyletic, therefore Candida species may be closely
related to various species that have a sexual state. A clade is a taxon, which is made of a genus containing species with a sexual state and
phylogenetically related Candida species.
The establishment of a taxonomic position of a definite species is best done with clades. A phylogenetic tree derived from various publications
by Kurtzman and his collaborators is shown below.
Back to top
Bibliography
Most of the information on the taxonomy of hemiascomycetous yeast can be found in the 5th edition of "The Yeast, a Taxonomical
study" by Kurtzman C.P., Boekhout T. and Fell J. (2011), Elsevier Amsterdam. The most recent changes in yeast taxonomy can be found in
Kurtzmann C.P. (2010), Phylogeny of the ascomycetous yeasts and the renaming of Pichia anomala to Wickerhamomyces anomalus. Antonie
Van Leeuwenhoek, 99: 13-23. The impact of genomics on yeast taxonomy has been recently reviewed by Casaregola S., Weiss S. and Morel G.
(2011), New perspectives in hemiascomycetous yeast taxonomy. C. R. Biol. Aug-Sep; (8-9):590-8. In this publication, all the changes of species
names since 1998 have also been reviewed.
Phylogeny.fr:
Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J.-F., Guindon S., Lefort V., Lescot M., Claverie J.-M.,
Gascuel O. Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Research. 2008 Jul 1; 36 (Web Server Issue):W465-9.
Epub 2008 Apr 19.
Back to top